
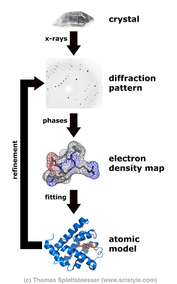
 The paper of (this) day was a meeting report about methods in protein crystallography. A perfect chance to repeat/learn some facts about X-Ray crystallography. X-ray crystallography is a method for determining the atomic structure of crystals. That helps to understand metals and minerals as well as organic and biological molecules. Every molecule which can be crystallised can be analysed by X-ray crystallography. The x-Rays diffract (change direction) because of the electric fields of the atoms in the crystal. By measuring the angles and intensities of the diffracted x-rays, the-dimensional electron-density models can be calculated. As the meeting report points out: the X-ray crystallography has two known bottlenecks: “the determination and refinement of three-dimensional structures subsequent to the wet-lab stage of obtaining suitable crystals of biological macromolecules.” The analysis of the diffraction pattern just works for pure crystalls with defined structure of the stable molecules. That is e.g. the problem with membrane proteins. Normally included in the phospho-lipid-bilayer, membrane proteins have hydrophil and hydrophob parts. That makes solvation and purification problematic. Moreover, the structure and stability is influenced by the surrounded lipids which may differ in the detergent compared to the original biological membrane. Therefore, the creation of pure crystals can be difficult. The second bottleneck refers to the problems in calculating a 3D electron density model based on the diffraction pattern. The diffraction pattern gives information about the angle and amplitude (intensity) of the diffraction. However, the phase information of the X-ray waves is lost. This is called the phase problem (see for example wikipedia article). Therefore a concrete model based on a single diffraction model is impossible, as different models would result in the same pattern of amplitudes and angles. However, comparing the diffraction pattern with the pattern of crystals made of similar molecule is one method to solve this problem. In this case computation models just have to explain the differences between both patterns instead of creating a 3d model from scratch. Unfortunately, the meeting report has no concrete conclusion. So I have to invent a message of the day: The next time you see the 3d structure of a molecule, especially membrane proteins, please respect the hard work which was spend in the progress of getting this knowledge. "Apotheosis, not apocalypse: methods in protein crystallography."
Victor S. Lamzin et al. Acta Crystallographica Section D: Biological Crystallography56.11 (2000): 1510-1511.
0 Kommentare
Hinterlasse eine Antwort. |
IdeaI love to increase my general science knowledge by reading papers from different fields of science. Here I share some of them. Archiv
März 2018
Kategorien
Alle
|
 RSS-Feed
RSS-Feed
